

Bệnh tan máu bẩm sinh (Thalassemia) là hội chứng rối loạn di truyền huyết học phổ biến.
Trong đó, Alpha Thalassemia đặc trưng bởi sự thiếu hụt chuỗi alpha-globin, khiến hồng cầu dễ bị phá hủy gây thiếu máu mãn tính. Việt Nam có tỷ lệ mang gen bệnh tương đối cao, đặc biệt ở vùng dân tộc thiểu số. Đáng lo ngại, nhiều người mang gen thể ẩn hoàn toàn khỏe mạnh, không có biểu hiện lâm sàng nhưng vẫn vô tình truyền gen bệnh sang cho thế hệ sau.
Bệnh alpha thalassemia là gì?
Huyết sắc tố (Hemoglobin – Hb) trong hồng cầu chịu trách nhiệm vận chuyển oxy đi nuôi cơ thể. Ở người trưởng thành, một phân tử huyết sắc tố bình thường được cấu tạo từ 2 chuỗi alpha ($alpha$) globin và 2 chuỗi beta ($beta$) globin.
Bệnh alpha thalassemia xảy ra khi có hiện tượng đột biến hoặc mất đoạn trên các gen quy định tổng hợp chuỗi alpha globin. Sự thiếu hụt chuỗi alpha khiến các chuỗi beta dư thừa tự liên kết thành các cấu trúc huyết sắc tố bất thường (như HbH hoặc Hb Bart’s). Những cấu trúc này làm màng hồng cầu trở nên lỏng lẻo, biến dạng và bị phá hủy sớm tại lách, dẫn đến tình trạng thiếu máu tan máu bẩm sinh.
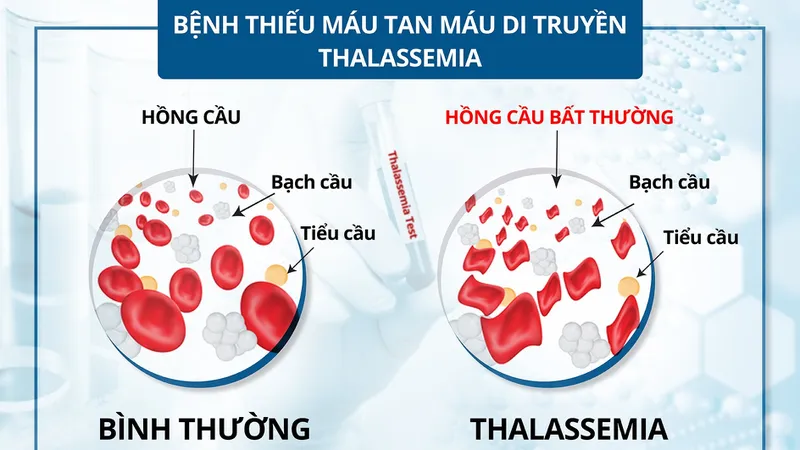
Bệnh alpha thalassemia là sự khiếm khuyết trong chu trình tổng hợp chuỗi alpha globin của tế bào hồng cầu (Nguồn: Sưu tầm)
Phân loại bệnh alpha thalassemia
Quá trình tổng hợp chuỗi alpha globin do cụm 4 gen trên cặp nhiễm sắc thể số 16 quy định (mỗi bên nhiễm sắc thể chứa 2 gen). Tùy vào số lượng gen bị tổn thương, bệnh được chia thành các thể sau:
Gen Triplicated Alpha
Cá thể sở hữu từ 5 đến 6 gen alpha do hiện tượng lặp đoạn. Thể này không có biểu hiện lâm sàng nhưng có thể làm tăng mức độ nghiêm trọng nếu đời con phối hợp với gen bệnh Beta Thalassemia.
Silent Carrier State (Thể Ẩn)
Chỉ có 1 trong số 4 gen bị tổn thương ($-alpha/alphaalpha$). Cơ thể vẫn sản xuất đủ lượng chuỗi alpha cần thiết. Người mang gen hoàn toàn khỏe mạnh, không thiếu máu và chỉ phát hiện được qua xét nghiệm DNA.
Alpha Thalassemia Trait
Có 2 trong số 4 gen bị tổn thương, gồm dạng dị hợp tử kép Cis ($–/alphaalpha$) hoặc đồng hợp tử Trans ($-alpha/-alpha$). Người bệnh chỉ bị thiếu máu rất nhẹ hoặc không có triệu chứng, hồng cầu nhỏ và nhạt màu.
Bệnh Hb H
Có 3 trong số 4 gen bị mất đoạn hoặc đột biến ($–/-alpha$). Lượng chuỗi alpha sụt giảm nghiêm trọng, hình thành huyết sắc tố bất thường HbH ($ beta_4 $). Bệnh nhân bị thiếu máu tan máu mức độ trung bình đến nặng, vàng da và lách to rõ rệt.
Hb Constant Spring (Hb CS)
Đây là dạng đột biến điểm tại bộ ba kết thúc của gen alpha. Chuỗi protein tạo ra dài hơn bình thường và cực kỳ kém ổn định, gây tan máu dai dẳng.
Homozygous Hb Constant Spring
Cả 2 gen alpha đều bị đột biến dạng Constant Spring. Người bệnh bị thiếu máu tan máu mức độ trung bình, hồng cầu biến dạng và tăng nguy cơ quá tải sắt.
Bệnh Hb H-Constant Spring
Thể phối hợp nặng thường gặp tại Việt Nam, xảy ra khi một nhiễm sắc thể mất cả 2 gen ($–$) và nhiễm sắc thể còn lại mang gen đột biến Constant Spring ($alpha^{CS}alpha$). Người bệnh bị thiếu máu nặng, biến dạng xương và phải truyền máu chu kỳ.
Alpha Thalassemia Major
Còn gọi là thể phù thai hoặc hội chứng Hb Bart’s, xảy ra khi toàn bộ 4 gen alpha bị mất đoạn ($–/–$). Thai nhi không thể tổng hợp chuỗi alpha, gây thiếu oxy và phù nề toàn thân nghiêm trọng. Thai thường bị lưu hoặc tử vong ngay sau sinh, đồng thời gây nguy cơ tiền sản giật lớn cho người mẹ.
Nguyên nhân gây bệnh alpha thalassemia
Alpha thalassemia di truyền theo quy luật di truyền lặn trên nhiễm sắc thể thường từ cha mẹ sang con cái, hoàn toàn không do tác động từ môi trường hay chế độ dinh dưỡng khi mang thai.
Nếu cả hai vợ chồng đều mang đặc tính Alpha Thalassemia thể nhẹ, xác suất trong mỗi lần sinh con sẽ là:
- 25% con bình thường (nhận 4 gen lành).
- 50% con mang gen thể nhẹ giống bố mẹ.
- 25% con mắc bệnh thể nặng (HbH hoặc phù thai Hb Bart’s).

Bệnh Thalassemia là một rối loạn máu di truyền (Nguồn: Sưu tầm)
Triệu chứng thalassemia alpha
Ở thể bệnh nặng (HbH hoặc phối hợp Constant Spring), các triệu chứng xuất hiện sớm và tiến triển rõ rệt:
- Mệt mỏi, ủ rũ, cáu gắt: Thiếu chuỗi alpha globin làm cấu trúc hemoglobin bị lỗi, giảm khả năng vận chuyển oxy. Cơ thể suy nhược, thiếu năng lượng khiến trẻ lờ đờ, kém tập trung và dễ quấy khóc.
- Hụt hơi: Do huyết sắc tố sụt giảm, cơ thể tăng tần suất hô hấp để bù oxy. Người bệnh dễ thở dốc, khó thở khi vận động, leo cầu thang hay tập thể thao.
- Nhịp tim nhanh: Tim phải co bóp nhanh, mạnh hơn để bù đắp lượng oxy thiếu hụt, gây hồi hộp, đánh trống ngực. Thiếu máu nặng kéo dài làm tim quá tải, dễ dẫn đến suy tim.
- Vàng da, vàng mắt: Hồng cầu dị dạng bị phá hủy hàng loạt (tán huyết) giải phóng lượng lớn bilirubin. Chất này tăng cao trong máu, tích tụ dưới da và kết mạc mắt gây vàng da, vàng mắt.
- Phát triển chậm, dậy thì muộn: Thiếu oxy và năng lượng khiến trẻ thấp còi, suy dinh dưỡng. Biến chứng ứ sắt làm suy tuyến yên, dẫn đến chậm phát triển thể chất và dậy thì muộn.
- Biến dạng xương mặt và đầu: Tủy xương tăng sản quá mức để bù máu làm xương bị giãn rộng, xốp và biến dạng. Biểu hiện rõ ở vùng mặt: trán dô, gò má cao, mũi tẹt, răng vẩu hàm trên.
- Dư thừa sắt trong cơ thể: Do tăng hấp thu sắt tại đường tiêu hóa và tích lũy từ lượng máu truyền định kỳ. Sắt dư thừa lắng đọng tại các mô, gây độc và hủy hoại cơ quan nội tạng.
- Tổn thương gan, tim và hệ thống nội tiết:
- Gan: Gây gan to, viêm gan mạn, xơ gan và suy gan.
- Tim: Gây bệnh cơ tim nhiễm sắt, rối loạn nhịp và suy tim sung huyết (nguyên nhân tử vong hàng đầu).
- Nội tiết: Tàn phá tuyến tụy gây đái tháo đường; tổn thương tuyến giáp và tuyến yên gây suy tuyến sinh dục, rối loạn tăng trưởng.
Biến chứng của bệnh alpha thalassemia
- Biến dạng xương: Tủy xương tăng sản quá mức để bù máu khiến lòng tủy phì đại và bào mòn lớp xương vỏ. Điều này làm biến dạng xương vùng đầu mặt, tạo nên “bộ mặt thalassemia” đặc trưng: trán dô, gò má cao, mũi tẹt và vẩu hàm trên.
- Thiếu máu: Do thiếu chuỗi alpha globin, cơ thể tạo ra hồng cầu nhỏ, lỗi và dễ vỡ. Lách tiêu hủy các hồng cầu này gây tán huyết mạn tính, làm suy kiệt oxy đến các cơ quan, gây mệt mỏi và suy tim do tim co bóp bù trừ. Thể nặng (phù thai) gây tử vong trước hoặc ngay sau sinh.
- Dị dạng xương: Quá trình tăng sản tủy gây dị dạng hệ xương vận động như ngực ức gà, vẹo cột sống, chân vòng kiềng. Xương bị mỏng, xốp và rất dễ gãy dù chỉ va chạm nhẹ, gây đau nhức mạn tính và hạn chế vận động.
- Bệnh tiểu đường: Sắt dư thừa từ hồng cầu vỡ và máu truyền tích tụ tại tuyến tụy. Chúng kích hoạt phản ứng oxy hóa phá hủy tế bào beta đảo tụy gây thiếu hụt insulin, dẫn đến đái tháo đường thứ phát và khiến việc điều trị trở nên phức tạp.
Phương pháp chẩn đoán và điều trị bệnh alpha thalassemia
Phương pháp chẩn đoán
- Tổng phân tích tế bào máu (CBC): Cho thấy tình trạng hồng cầu nhỏ (MCV
- Định lượng Ferritin: Đánh giá lượng sắt dự trữ để phân biệt với thiếu máu thiếu sắt.
- Điện di huyết sắc tố: Tìm kiếm sự xuất hiện của các huyết sắc tố bất thường như HbH.
- Xét nghiệm di truyền DNA: Tiêu chuẩn vàng để xác định chính xác kiểu đột biến gen.
Phương pháp điều trị
- Truyền máu: Truyền hồng cầu lắng định kỳ để duy trì nồng độ Hb ổn định từ 9.5 – 10 g/dl.
- Thải sắt: Sử dụng thuốc thải sắt khi chỉ số Ferritin vượt quá 1.000 ng/ml nhằm bảo vệ các cơ quan nội tạng.
- Bổ sung Acid Folic: Cung cấp nguyên liệu tạo hồng cầu. Tuyệt đối không dùng thuốc bổ sung sắt.
- Cắt lách: Cân nhắc khi lách quá to hoặc xảy ra hiện tượng cường lách làm tăng nhu cầu truyền máu.
- Ghép tế bào gốc: Phương pháp duy nhất có thể chữa khỏi hoàn toàn bệnh nhưng chi phí cao và đòi hỏi người hiến phù hợp HLA.

Hiểu rõ tiền sử di truyền và thực hiện các xét nghiệm trước khi kết hôn là cách tốt nhất để phòng tránh bệnh (Nguồn: Sưu tầm)
Biện pháp phòng ngừa bệnh alpha thalassemia
- Sàng lọc tiền hôn nhân: Xét nghiệm công thức máu và phân tích DNA trước khi kết hôn để phát hiện người mang gen lành.
- Chẩn đoán trước sinh: Chọc hút gai nhau (tuần thai 11 – 14) hoặc chọc dò dịch ối (tuần thai 16 – 20) để xác định kiểu gen của thai nhi.
- Chẩn đoán di truyền tiền làm tổ (PGD): Thực hiện thụ tinh trong ống nghiệm (IVF) và sàng lọc phôi khỏe mạnh trước khi chuyển vào tử cung.
Những câu hỏi thường gặp về alpha thalassemia
Alpha thalassemia dị hợp là gì?
Là trạng thái cơ thể chỉ mang 1 hoặc 2 gen alpha bị đột biến. Người mang gen dị hợp hoàn toàn khỏe mạnh, không có triệu chứng lâm sàng và chỉ có ý nghĩa về mặt di truyền cho đời sau.
Thiếu máu khi mang thai có nguy hiểm không?
Rất nguy hiểm. Người mẹ mang gen bệnh dễ bị sảy thai, sinh non hoặc băng huyết. Nếu thai nhi mắc thể nặng (phù thai Hb Bart’s), thai sẽ bị suy tim nặng, phù nề và thường bị lưu trong bụng mẹ.
Chăm sóc người bệnh alpha thalassemia như thế nào?
- Dinh dưỡng: Ăn uống cân bằng, bổ sung acid folic. Hạn chế thực phẩm giàu sắt (thịt đỏ) và uống nước chè xanh sau ăn để giảm hấp thu sắt.
- Tránh dùng thuốc chứa sắt: Tuyệt đối không tự ý uống các loại thuốc bổ máu chứa sắt.
- Phòng ngừa nhiễm trùng: Tiêm phòng đầy đủ các loại vắc xin (cúm, phế cầu, viêm màng não) và giữ gìn vệ sinh sạch sẽ.
- Vận động: Tập thể dục nhẹ nhàng (đi bộ, yoga), tránh các môn thể thao đối kháng va đập mạnh.
Tóm lại, Alpha Thalassemia là hội chứng rối loạn huyết học di truyền phức tạp, để lại hệ lụy nặng nề cho sức khỏe. Tuy nhiên, nếu chủ động thực hiện tốt công tác sàng lọc tiền hôn nhân, tư vấn di truyền kịp thời và tuân thủ đúng phác đồ điều trị y khoa hiện đại, chúng ta hoàn toàn có thể ngăn chặn sự tiếp diễn của gen bệnh. Việc này giúp mang lại cuộc sống khỏe mạnh, trọn vẹn cho thế hệ tương lai và giảm bớt gánh nặng cho xã hội.
Nguồn tham khảo:
Johns Hopkins Medicine:
https://www.hopkinsmedicine.org/health/conditions-and-diseases/alpha-thalassemia
National Library of Medicine:
https://www.hopkinsmedicine.org/health/conditions-and-diseases/alpha-thalassemia
📝Nguồn tài liệu: Chọn lọc từ nhiều nguồn y tế uy tín
🔎Lưu ý: Bài viết chỉ nhằm mục đích cung cấp thông tin tổng quan. Vui lòng tham khảo ý kiến của Bác sĩ, Dược sĩ hoặc chuyên gia y tế để nhận được hướng dẫn phù hợp với tình trạng sức khỏe của bạn.