
Bệnh Niemann-Pick là một rối loạn di truyền hiếm gặp ảnh hưởng nghiêm trọng đến khả năng phân giải và vận chuyển chất béo trong tế bào, gây ra tình trạng tích tụ lipid nội bào dẫn đến tổn thương gan, lách, hệ thần kinh và nhiều cơ quan khác. Đây là một căn bệnh tuy ít phổ biến nhưng lại có ảnh hưởng nặng nề đến sức khỏe và tuổi thọ của người bệnh nếu không được phát hiện và điều trị kịp thời.

Theo các nghiên cứu được đăng tải trên National Organization for Rare Disorders (NORD), tỷ lệ mắc bệnh Niemann-Pick rơi vào khoảng 1/250.000 trẻ sơ sinh, và tỷ lệ này có thể thay đổi tùy vào khu vực địa lý và yếu tố di truyền trong cộng đồng. Với sự phát triển của y học hiện đại, việc hiểu biết rõ về cơ chế, triệu chứng và phương pháp điều trị Niemann-Pick ngày càng trở nên cần thiết — không chỉ với đội ngũ y tế mà còn với cộng đồng và các gia đình có nguy cơ cao.
1. Bệnh Niemann-Pick là gì?
Niemann-Pick là tên gọi chung của một nhóm bệnh di truyền liên quan đến sự rối loạn trong quá trình chuyển hóa lipid, đặc biệt là sphingomyelin và cholesterol. Bệnh thuộc nhóm bệnh rối loạn tích trữ lysosome (lysosomal storage disorders – LSDs), tức là các chất béo không thể được phân giải bình thường mà tích tụ trong lysosome – một bào quan quan trọng trong tế bào.
Được đặt tên theo hai nhà khoa học là Albert Niemann và Ludwig Pick – những người đầu tiên mô tả căn bệnh này vào đầu thế kỷ 20, Niemann-Pick hiện được chia làm nhiều thể loại khác nhau (A, B, C), mỗi loại có đặc điểm bệnh sinh và biểu hiện lâm sàng riêng biệt. Tuy nhiên, điểm chung là đều dẫn đến hậu quả tổn thương tế bào nghiêm trọng.
2. Cơ chế bệnh sinh và nguyên nhân
2.1. Vai trò của enzyme sphingomyelinase và protein NPC1/NPC2
Ở người khỏe mạnh, enzyme sphingomyelinase acid (ASM) có chức năng phân giải sphingomyelin – một loại lipid có trong màng tế bào. Khi gen SMPD1 (mã hóa enzyme ASM) bị đột biến, enzyme này hoạt động không hiệu quả, dẫn đến việc sphingomyelin tích tụ trong các tế bào của gan, lách, phổi và hệ thần kinh trung ương.
Đối với loại C của bệnh Niemann-Pick, đột biến xảy ra ở các gen NPC1 hoặc NPC2 – vốn liên quan đến quá trình vận chuyển cholesterol và glycosphingolipid trong tế bào. Khi các protein này hoạt động sai lệch, cholesterol bị mắc kẹt trong lysosome, gây nhiễm độc tế bào và rối loạn chức năng cơ quan.
2.2. Di truyền học của bệnh Niemann-Pick
Đây là bệnh di truyền lặn trên nhiễm sắc thể thường, nghĩa là một người cần phải nhận 2 bản sao đột biến (một từ mỗi bố mẹ) để phát triển bệnh. Những người chỉ mang một bản sao đột biến gọi là “người mang gen” và thường không có triệu chứng.
Các gen thường liên quan bao gồm:
- SMPD1 – gây ra loại A và B.
- NPC1 và NPC2 – gây ra loại C.
Đặc biệt, bệnh Niemann-Pick loại A và B có thể được phát hiện sớm ở trẻ sơ sinh thông qua tầm soát enzyme, trong khi loại C thường chẩn đoán muộn do triệu chứng diễn tiến chậm và khó phát hiện hơn.
3. Phân loại bệnh Niemann-Pick
3.1. Loại A
Loại A là dạng nghiêm trọng nhất, khởi phát từ giai đoạn sơ sinh với triệu chứng tiến triển nhanh chóng. Trẻ thường bị gan lách to, chậm phát triển trí tuệ và thể chất, kèm theo các biểu hiện thần kinh rõ rệt. Hầu hết bệnh nhân loại A tử vong trước 3 tuổi.
3.2. Loại B
Loại B thường nhẹ hơn, không ảnh hưởng đến thần kinh trung ương. Bệnh nhân có thể sống đến tuổi trưởng thành, nhưng vẫn phải đối mặt với các vấn đề gan lách to, rối loạn lipid máu và nguy cơ xơ gan, suy hô hấp. Loại này hiện được nghiên cứu nhiều trong liệu pháp thay thế enzyme.
3.3. Loại C
Đây là dạng bệnh phổ biến nhất trong nhóm Niemann-Pick, do đột biến gen NPC1 hoặc NPC2 gây rối loạn vận chuyển cholesterol trong tế bào. Triệu chứng thường xuất hiện muộn – từ thời thơ ấu đến tuổi trưởng thành – và ảnh hưởng nặng đến thần kinh: sa sút trí tuệ, mất điều hòa, khó nuốt, rối loạn vận động nhãn cầu.
3.3.1. So sánh các loại Niemann-Pick
| Đặc điểm | Loại A | Loại B | Loại C |
|---|---|---|---|
| Gen liên quan | SMPD1 | SMPD1 | NPC1/NPC2 |
| Khởi phát | Sơ sinh | Trẻ em | Trẻ em/trưởng thành |
| Ảnh hưởng thần kinh | Rất nặng | Không | Nặng, tiến triển |
| Tuổi thọ | ≤ 3 tuổi | Đến tuổi trưởng thành | Biến thiên |
4. Triệu chứng và dấu hiệu nhận biết
4.1. Triệu chứng toàn thân
- Gan và lách to bất thường, gây bụng chướng
- Chậm phát triển thể chất
- Dễ nhiễm trùng do suy giảm miễn dịch
4.2. Triệu chứng thần kinh
Thường gặp ở loại A và C, bao gồm:
- Sa sút trí tuệ
- Co giật
- Mất điều hòa vận động
- Khó nuốt, sặc thức ăn
4.3. Dấu hiệu đặc trưng khác
- Chuyển động mắt dọc chậm (vertical supranuclear gaze palsy)
- Bất thường hành vi
- Suy giảm khả năng học tập
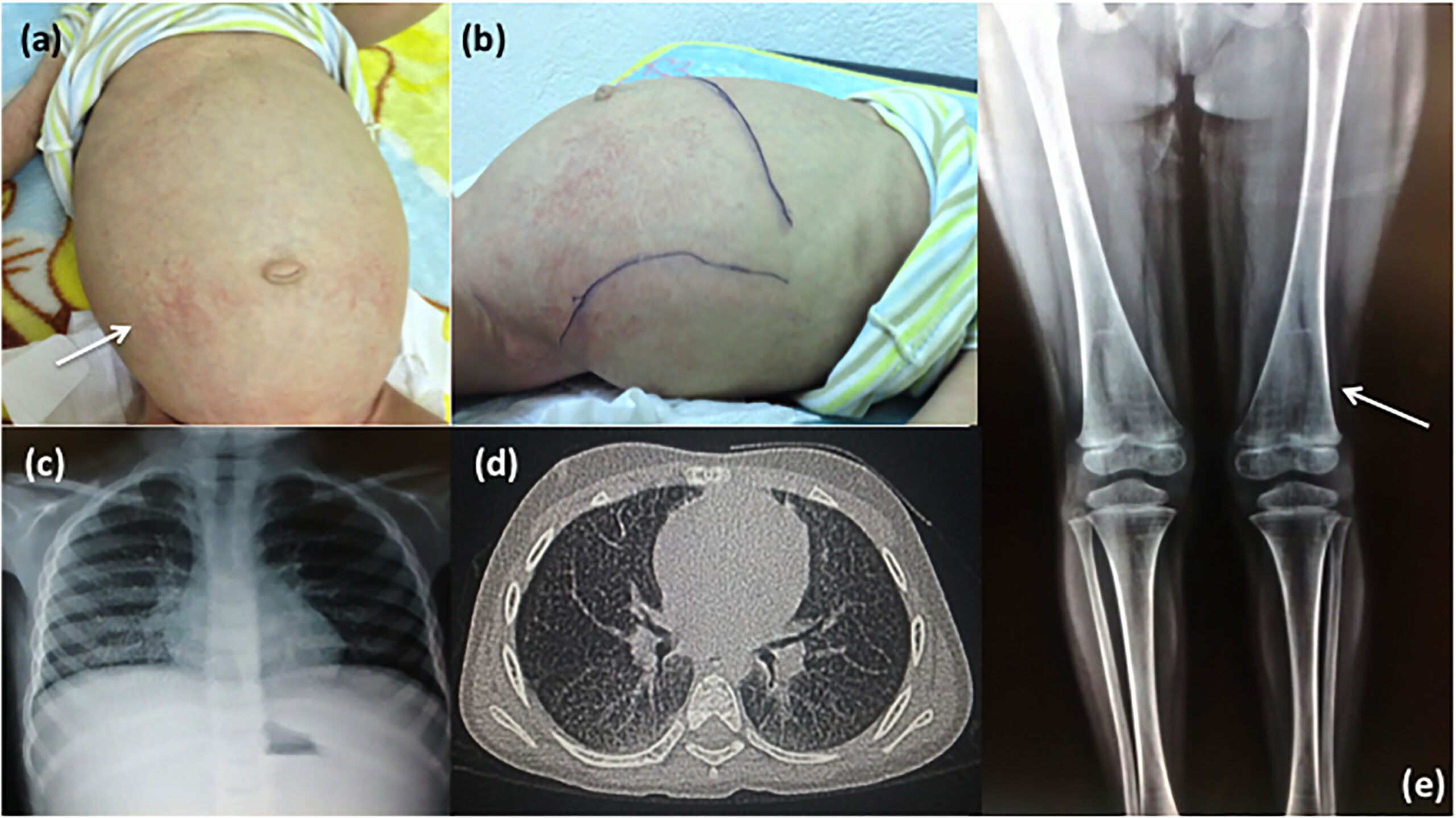
5. Chẩn đoán bệnh Niemann-Pick
5.1. Xét nghiệm enzyme và sinh học phân tử
Chẩn đoán bệnh Niemann-Pick thường bắt đầu từ các dấu hiệu lâm sàng nghi ngờ, sau đó được xác nhận bằng các xét nghiệm chuyên sâu. Trong trường hợp loại A và B, xét nghiệm định lượng enzyme sphingomyelinase acid (ASM) trong máu hoặc tế bào nuôi cấy giúp xác định sự thiếu hụt enzyme.
Với loại C, phương pháp hiệu quả nhất là phân tích DNA để phát hiện các đột biến trên gen NPC1 hoặc NPC2. Ngoài ra, một số kỹ thuật như test filipin staining trong nuôi cấy tế bào da cũng được sử dụng để kiểm tra sự tích tụ cholesterol trong tế bào.
5.2. Sinh thiết tế bào
Ở một số trường hợp, bác sĩ có thể chỉ định sinh thiết mô (thường là tủy xương hoặc gan) để quan sát dưới kính hiển vi. Nếu phát hiện sự xuất hiện của các “tế bào bọt” – dạng tế bào phì đại với các túi chứa lipid không phân giải được – thì có thể củng cố chẩn đoán bệnh.
5.3. Chẩn đoán trước sinh
Đối với các gia đình có tiền sử mắc bệnh Niemann-Pick, việc tư vấn di truyền và chẩn đoán trước sinh là vô cùng quan trọng. Xét nghiệm ADN phôi thai bằng phương pháp chọc ối hoặc sinh thiết gai nhau có thể giúp phát hiện gen đột biến từ sớm, từ đó đưa ra quyết định phù hợp cho thai kỳ.
5.3.1. Các dấu hiệu gợi ý cần xét nghiệm
- Tiền sử gia đình có người mắc bệnh Niemann-Pick
- Trẻ sơ sinh có gan, lách to không rõ nguyên nhân
- Chậm phát triển trí tuệ và vận động, kèm theo rối loạn thần kinh
6. Phương pháp điều trị hiện nay
6.1. Điều trị triệu chứng và hỗ trợ
Hiện nay, không có phương pháp chữa khỏi hoàn toàn bệnh Niemann-Pick, nhưng các biện pháp điều trị triệu chứng giúp cải thiện chất lượng sống đáng kể. Những biện pháp hỗ trợ bao gồm:
- Vật lý trị liệu: giúp duy trì chức năng vận động
- Chế độ ăn uống: giàu calo, hỗ trợ tiêu hóa
- Điều trị co giật và rối loạn hành vi: bằng thuốc
- Chăm sóc hỗ trợ hô hấp: đặc biệt ở giai đoạn cuối
6.2. Điều trị đặc hiệu cho loại C: Miglustat
Miglustat là loại thuốc duy nhất hiện nay được chấp thuận ở một số quốc gia để điều trị bệnh Niemann-Pick loại C. Thuốc này ức chế enzyme liên quan đến quá trình tích tụ glycosphingolipid, giúp làm chậm tiến triển của bệnh, đặc biệt là các triệu chứng thần kinh.
Theo một nghiên cứu được công bố trên Journal of Inherited Metabolic Disease, Miglustat giúp cải thiện chức năng nuốt và giảm tốc độ mất chức năng vận động ở một số bệnh nhân nhi. Tuy nhiên, tác dụng không giống nhau với tất cả trường hợp và cần được chỉ định bởi bác sĩ chuyên khoa.
6.3. Nghiên cứu và liệu pháp tương lai
Nhiều hướng nghiên cứu đang được triển khai để tìm ra liệu pháp điều trị căn cơ cho bệnh Niemann-Pick, bao gồm:
- Liệu pháp thay thế enzyme (ERT): đang được thử nghiệm lâm sàng cho loại B
- Liệu pháp gen (Gene therapy): nhằm sửa chữa đột biến gen SMPD1 hoặc NPC
- Điều trị bằng tế bào gốc: phục hồi tổn thương mô
Mặc dù chưa có kết quả chính thức, nhưng các bước tiến trong lĩnh vực này mở ra hy vọng mới cho bệnh nhân và gia đình.
7. Tiên lượng và biến chứng
7.1. Tuổi thọ theo từng loại bệnh
| Loại bệnh | Tuổi thọ trung bình |
|---|---|
| Loại A | 2 – 3 tuổi |
| Loại B | Trưởng thành (nhiều người sống >40 tuổi) |
| Loại C | Biến thiên, phụ thuộc vào tuổi khởi phát |
7.2. Các biến chứng nguy hiểm
- Suy hô hấp do viêm phổi hoặc yếu cơ hô hấp
- Co giật khó kiểm soát
- Suy dinh dưỡng nặng
- Sa sút trí tuệ không hồi phục
- Tử vong sớm ở thể nặng
8. Câu chuyện thực tế: Hy vọng trong cuộc chiến với Niemann-Pick
“Bé Linh, được chẩn đoán mắc bệnh Niemann-Pick loại C khi mới 1 tuổi rưỡi. Dù trải qua nhiều lần nhập viện vì viêm phổi, co giật và khó nuốt, gia đình vẫn kiên trì điều trị bằng Miglustat, kết hợp vật lý trị liệu và chăm sóc dinh dưỡng chuyên biệt. Hiện tại, bé đã 5 tuổi và vẫn đang tiếp tục chiến đấu mỗi ngày. Câu chuyện của bé là minh chứng cho nghị lực và hy vọng – dù bệnh hiếm nhưng không phải là dấu chấm hết.”
9. Kết luận
Bệnh Niemann-Pick là một thách thức y học với nhiều biến thể phức tạp, nhưng việc hiểu đúng và phát hiện sớm có thể giúp cải thiện đáng kể chất lượng cuộc sống cho người bệnh. Dù hiện chưa có phương pháp điều trị dứt điểm, các liệu pháp hỗ trợ và thuốc đặc hiệu như Miglustat đã mang lại hy vọng cho nhiều gia đình.
Quan trọng hơn hết, cộng đồng cần có sự đồng cảm và hỗ trợ đối với các bệnh hiếm, đồng thời tăng cường nhận thức để tư vấn di truyền và chẩn đoán trước sinh được thực hiện sớm và hiệu quả.
10. Câu hỏi thường gặp (FAQ)
1. Bệnh Niemann-Pick có di truyền không?
Có. Đây là bệnh di truyền lặn trên nhiễm sắc thể thường. Bệnh chỉ biểu hiện khi cả cha và mẹ đều mang gen bệnh.
2. Làm sao để phát hiện sớm bệnh Niemann-Pick?
Phát hiện qua xét nghiệm enzyme, sinh học phân tử, chẩn đoán di truyền, đặc biệt quan trọng trong gia đình có tiền sử bệnh.
3. Có thể điều trị khỏi bệnh Niemann-Pick không?
Hiện tại chưa có phương pháp chữa khỏi hoàn toàn. Tuy nhiên, việc điều trị triệu chứng và dùng thuốc như Miglustat có thể giúp làm chậm tiến triển bệnh.
4. Người mang gen bệnh có biểu hiện triệu chứng không?
Không. Người mang gen bệnh chỉ có 1 bản sao gen đột biến nên không biểu hiện triệu chứng, nhưng có thể truyền bệnh cho con cái.
5. Bệnh Niemann-Pick có phổ biến không?
Không. Đây là bệnh hiếm gặp với tỷ lệ khoảng 1/250.000 – 1/150.000 ca sinh sống, tùy từng thể bệnh và khu vực địa lý.
📝Nguồn tài liệu: Chọn lọc từ nhiều nguồn y tế uy tín
🔎Lưu ý: Bài viết chỉ nhằm mục đích cung cấp thông tin tổng quan. Vui lòng tham khảo ý kiến của Bác sĩ, Dược sĩ hoặc chuyên gia y tế để nhận được hướng dẫn phù hợp với tình trạng sức khỏe của bạn.