
Hội chứng Bardet-Biedl (BBS) là một rối loạn di truyền hiếm gặp nhưng có sức ảnh hưởng mạnh mẽ đến nhiều hệ cơ quan trong cơ thể. Từ các biểu hiện rõ ràng như béo phì, suy giảm thị lực đến những ảnh hưởng tiềm ẩn như suy thận, rối loạn nội tiết và phát triển trí tuệ – BBS chính là minh chứng rõ nét cho tầm quan trọng của chẩn đoán và theo dõi bệnh di truyền sớm. Trong bài viết này, chúng tôi sẽ giúp bạn tìm hiểu toàn diện về căn bệnh này, từ nguyên nhân, dấu hiệu nhận biết, phương pháp chẩn đoán đến các hướng điều trị hiện đại và hiệu quả nhất.

1. Hội chứng Bardet-Biedl là gì?
Hội chứng Bardet-Biedl là một rối loạn di truyền lặn trên nhiễm sắc thể thường, thuộc nhóm các bệnh do rối loạn chức năng của lông mao tế bào (cilia). Đây là các cấu trúc siêu nhỏ có vai trò quan trọng trong sự phát triển, dẫn truyền tín hiệu và duy trì hoạt động bình thường của nhiều cơ quan trong cơ thể.
Hội chứng này thường biểu hiện qua rối loạn thị giác, béo phì, bất thường chi, rối loạn thận, sinh sản và phát triển trí tuệ. Mặc dù rất hiếm, với tỷ lệ mắc khoảng 1/140.000 – 1/160.000 người tại Bắc Mỹ và Châu Âu, nhưng tỷ lệ có thể cao hơn tại các cộng đồng có tần suất kết hôn cận huyết.
Theo Trung tâm Y khoa Quốc gia Hoa Kỳ (NIH), hiện đã xác định được ít nhất 21 gen (BBS1 đến BBS21) liên quan đến hội chứng Bardet-Biedl, trong đó gen BBS1 và BBS10 là phổ biến nhất.
2. Nguyên nhân gây hội chứng Bardet-Biedl
Hội chứng Bardet-Biedl có cơ chế di truyền lặn trên nhiễm sắc thể thường. Điều này có nghĩa là người bệnh phải nhận một bản sao gen đột biến từ cả cha lẫn mẹ thì mới biểu hiện triệu chứng bệnh.
2.1. Vai trò của các gen BBS
Ít nhất 21 gen đã được xác định có liên quan đến hội chứng này, trong đó các gen thường gặp bao gồm:
- BBS1: liên quan đến khoảng 23% các trường hợp
- BBS10: chiếm khoảng 20%
- BBS2, BBS12, BBS9: cũng có thể gây bệnh
Các gen này chịu trách nhiệm mã hóa protein hình thành nên cấu trúc và chức năng của lông mao – một thành phần tế bào đóng vai trò quan trọng trong phát tín hiệu và vận động của tế bào.
2.2. Cơ chế bệnh sinh
Khi các gen này bị đột biến, quá trình tạo và duy trì lông mao bị ảnh hưởng, từ đó dẫn đến rối loạn chức năng ở nhiều cơ quan như mắt, thận, não, tuyến nội tiết và cơ quan sinh sản.
Các nghiên cứu cho thấy lông mao là bộ máy cảm biến của tế bào. Khi lông mao không hoạt động đúng cách, cơ thể sẽ rối loạn trong việc định hướng phát triển mô, truyền tín hiệu nội bào và hoạt động điều hòa nội tiết.
3. Triệu chứng lâm sàng phổ biến
Hội chứng Bardet-Biedl là một rối loạn đa hệ, biểu hiện triệu chứng rất đa dạng và thường bắt đầu xuất hiện từ thời thơ ấu. Các triệu chứng dưới đây là điển hình:
3.1. Thoái hóa võng mạc
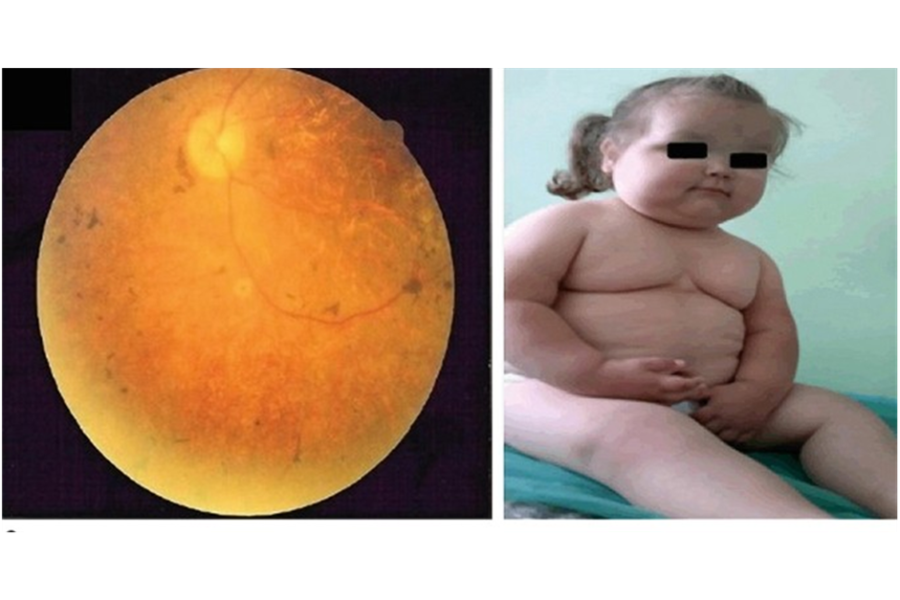
Đây là triệu chứng sớm và rõ nét nhất. Bệnh nhân thường bắt đầu có biểu hiện quáng gà (khó nhìn trong bóng tối) từ thời thơ ấu và dần dần giảm thị lực nghiêm trọng theo tuổi. Dạng bệnh phổ biến là viêm võng mạc sắc tố, có thể dẫn đến mù lòa ở tuổi trưởng thành.
3.2. Béo phì khởi phát sớm
Trẻ em mắc BBS thường bị béo phì trung tâm (bụng to, tích mỡ vùng eo và ngực) ngay từ giai đoạn đầu đời, không liên quan đến chế độ ăn uống. Nguyên nhân do rối loạn nội tiết và mất kiểm soát cảm giác no.
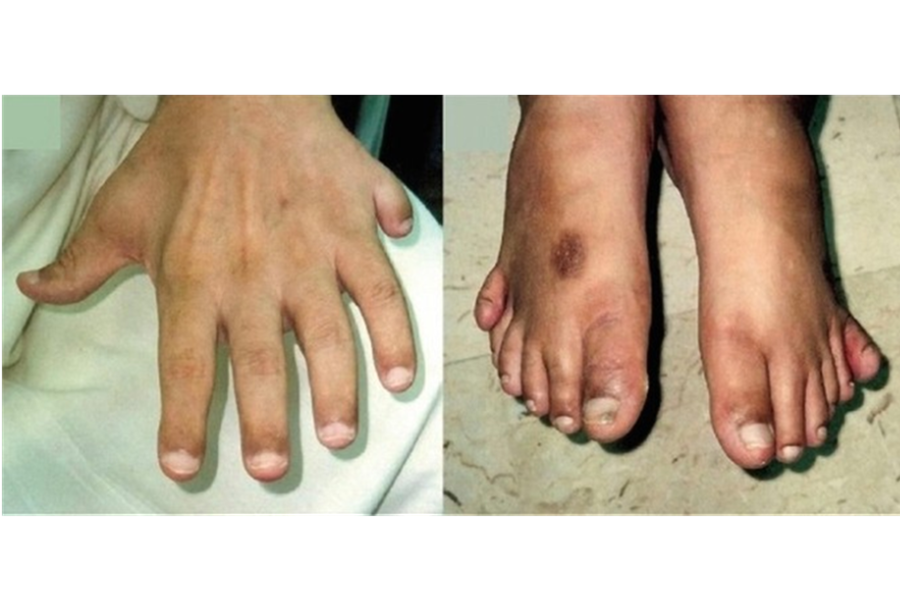
3.3. Đa ngón và dị tật chi
Khoảng 70–80% bệnh nhân có đa ngón tay hoặc ngón chân, đặc biệt là ngón thừa ở cạnh ngoài bàn tay hoặc bàn chân. Một số trường hợp bị ngắn chi hoặc dị dạng nhẹ ở tay chân.
3.4. Suy giảm chức năng thận
Hệ tiết niệu – đặc biệt là thận – thường bị ảnh hưởng nghiêm trọng. Có thể bao gồm:
- Thận nhỏ, kém phát triển
- Giãn bể thận, nhiễm trùng niệu tái phát
- Suy thận mạn, cần lọc máu hoặc ghép thận
3.5. Rối loạn sinh sản
Nam giới thường có tinh hoàn nhỏ, tinh trùng yếu hoặc vô tinh. Phụ nữ có thể bị buồng trứng đa nang, kinh nguyệt không đều, vô sinh.
3.6. Trí tuệ chậm phát triển
Đa số bệnh nhân có mức IQ trung bình thấp hoặc rối loạn phát triển trí tuệ mức nhẹ đến trung bình. Một số có biểu hiện tăng động, giảm khả năng tập trung hoặc rối loạn phổ tự kỷ.
4. Các dấu hiệu khác đi kèm
Bên cạnh các triệu chứng chính, người mắc hội chứng Bardet-Biedl có thể có một số biểu hiện khác như:
- Dị tật tim bẩm sinh: bao gồm thông liên nhĩ, thông liên thất, bất thường van tim
- Rối loạn nội tiết: đái tháo đường type 2, giảm hormone tăng trưởng, suy giáp
- Rối loạn tiêu hóa: táo bón mãn tính, gan nhiễm mỡ
- Thiểu sản men răng: dễ bị sâu răng, hỏng men, hở hàm ếch
Không phải tất cả các bệnh nhân đều có cùng triệu chứng. Mức độ và loại biểu hiện phụ thuộc vào gen đột biến cụ thể cũng như mức độ ảnh hưởng của từng cá nhân.
5. Chẩn đoán hội chứng Bardet-Biedl như thế nào?
Chẩn đoán hội chứng BBS là một quá trình kết hợp giữa khai thác lâm sàng, tiền sử gia đình và xét nghiệm di truyền hiện đại.
5.1. Khám lâm sàng và tiền sử gia đình
Việc ghi nhận các triệu chứng lâm sàng đặc trưng và xác định những trường hợp tương tự trong gia đình giúp gợi ý hướng chẩn đoán di truyền.
5.2. Xét nghiệm di truyền
Xét nghiệm giải trình tự gen (gene panel hoặc exome sequencing) để phát hiện các đột biến trong các gen BBS.
- Phân tích gen BBS1, BBS10 trước do phổ biến
- Nếu không phát hiện, xét nghiệm toàn bộ các gen liên quan
5.3. Đánh giá chức năng cơ quan
Sau khi nghi ngờ hoặc xác định chẩn đoán, cần tiến hành các cận lâm sàng để đánh giá tổn thương cơ quan:
- Khám mắt: đo thị lực, chụp OCT võng mạc
- Siêu âm, MRI thận: đánh giá cấu trúc thận
- Xét nghiệm hormone: đánh giá tuyến giáp, hormone sinh dục
- Khám thần kinh, tâm lý học: kiểm tra khả năng học tập, hành vi
6. Phân biệt với các bệnh lý tương tự
Do hội chứng Bardet-Biedl có nhiều triệu chứng tương đồng với các bệnh di truyền khác, việc phân biệt rõ ràng là rất quan trọng trong chẩn đoán và điều trị.
| Bệnh lý | Đặc điểm giống | Đặc điểm khác biệt |
|---|---|---|
| Hội chứng Alström | Béo phì, thoái hóa võng mạc, tiểu đường | Không có dị tật chi, có điếc cảm giác thần kinh |
| Hội chứng Laurence-Moon | Giảm thị lực, chậm phát triển | Không có đa ngón, thường có liệt chi dưới |
Chẩn đoán phân biệt dựa trên các đặc điểm lâm sàng, hình ảnh học, cũng như kết quả xét nghiệm gen là yếu tố then chốt để tránh sai sót trong điều trị.
7. Hướng điều trị và can thiệp hỗ trợ
Hiện nay, hội chứng Bardet-Biedl chưa có phương pháp điều trị khỏi hoàn toàn. Mục tiêu điều trị chủ yếu là kiểm soát triệu chứng, ngăn ngừa biến chứng và nâng cao chất lượng sống cho người bệnh.
7.1. Điều trị triệu chứng
- Béo phì: áp dụng chế độ ăn kiêng, luyện tập, kết hợp can thiệp nội tiết nếu cần
- Thoái hóa võng mạc: sử dụng thiết bị hỗ trợ thị giác, môi trường ánh sáng phù hợp
- Suy thận: theo dõi chỉ số chức năng thận định kỳ, can thiệp sớm, lọc máu hoặc ghép thận
7.2. Hỗ trợ thị giác và thiết bị trợ giúp
Người bệnh cần được hướng dẫn sử dụng thiết bị hỗ trợ như gậy thông minh, kính phóng đại, hệ thống định vị âm thanh để hỗ trợ sinh hoạt độc lập.
7.3. Theo dõi chức năng thận và hormone
- Đo creatinine máu, tổng phân tích nước tiểu
- Đánh giá nồng độ hormone tuyến giáp, hormone sinh dục định kỳ
7.4. Tư vấn di truyền và hỗ trợ gia đình
Vì đây là bệnh lý di truyền, cần tư vấn cho cha mẹ, người thân về nguy cơ tái phát nếu có kế hoạch sinh thêm con. Tư vấn di truyền giúp phòng tránh và chủ động tầm soát bệnh trong cộng đồng có nguy cơ cao.
8. Tiên lượng bệnh nhân Bardet-Biedl
Tiên lượng của bệnh nhân phụ thuộc vào mức độ ảnh hưởng của các cơ quan, đặc biệt là chức năng thị giác và thận. Những người được chẩn đoán sớm và quản lý tốt có thể duy trì chất lượng sống tương đối ổn định.
- Biến chứng nguy hiểm: mù lòa, suy thận mạn giai đoạn cuối, tiểu đường, vô sinh
- Chất lượng sống: cải thiện nếu có chương trình hỗ trợ xã hội, giáo dục đặc biệt
Với sự tiến bộ của y học, nhiều bệnh nhân có thể học tập, làm việc và sống độc lập nhờ các biện pháp hỗ trợ phù hợp.
9. Làm gì khi gia đình có người mắc hội chứng Bardet-Biedl?
Nếu trong gia đình có người mắc bệnh, cần:
- Thực hiện xét nghiệm gen cho các thành viên: xác định người mang gen lặn để phòng tránh di truyền cho thế hệ sau.
- Tham gia nhóm hỗ trợ: kết nối với các tổ chức hỗ trợ bệnh nhân BBS, nhóm Facebook hoặc cộng đồng trực tuyến để chia sẻ kinh nghiệm.
- Tư vấn trước sinh: sử dụng công nghệ sàng lọc trước sinh nếu có ý định sinh thêm con.
10. Tổng kết
Hội chứng Bardet-Biedl là một rối loạn di truyền hiếm nhưng ảnh hưởng sâu rộng đến sức khỏe và chất lượng sống của người bệnh. Việc nhận diện sớm các dấu hiệu lâm sàng, kết hợp với chẩn đoán di truyền chính xác và can thiệp toàn diện, sẽ mang lại cơ hội cải thiện đáng kể cho bệnh nhân và gia đình.
Y học hiện đại đang tiến gần hơn đến việc cá nhân hóa điều trị các bệnh di truyền như BBS, mở ra hi vọng mới trong tương lai.
Câu hỏi thường gặp (FAQ)
1. Hội chứng Bardet-Biedl có chữa khỏi được không?
Hiện chưa có phương pháp điều trị khỏi hoàn toàn. Tuy nhiên, kiểm soát triệu chứng hiệu quả có thể giúp người bệnh sống khỏe mạnh và độc lập.
2. Bệnh có di truyền cho con không?
Có. Nếu cả cha và mẹ đều mang gen đột biến thì khả năng truyền bệnh cho con là 25%. Việc tư vấn di truyền là rất quan trọng.
3. Bệnh có thể phát hiện từ khi nào?
Một số dấu hiệu có thể nhận ra ngay từ sơ sinh (đa ngón, béo phì), nhưng đa phần được phát hiện trong giai đoạn trẻ học mẫu giáo hoặc tiểu học khi thị lực giảm dần.
4. Có thể mang thai an toàn nếu mắc BBS không?
Phụ nữ mắc BBS vẫn có thể mang thai nhưng cần được theo dõi sát về nội tiết, thận và các nguy cơ biến chứng. Cần tư vấn kỹ trước khi có thai.
5. Ở đâu có thể xét nghiệm gen để phát hiện BBS?
Các bệnh viện lớn như Bệnh viện Từ Dũ, Vinmec, Bệnh viện Đại học Y Dược TP.HCM, Bệnh viện Nhi Trung Ương, và các trung tâm xét nghiệm di truyền như Genetica, Chekco… đều có cung cấp dịch vụ xét nghiệm di truyền BBS.
Hãy hành động ngay hôm nay
Nếu bạn nghi ngờ người thân hoặc con em mình có dấu hiệu mắc hội chứng Bardet-Biedl, đừng chần chừ. Hãy đặt lịch khám tại các cơ sở y tế uy tín, thực hiện xét nghiệm di truyền và nhận tư vấn chuyên sâu từ các chuyên gia.
Chẩn đoán sớm – Can thiệp đúng – Sống khỏe mỗi ngày!
📝Nguồn tài liệu: Chọn lọc từ nhiều nguồn y tế uy tín
🔎Lưu ý: Bài viết chỉ nhằm mục đích cung cấp thông tin tổng quan. Vui lòng tham khảo ý kiến của Bác sĩ, Dược sĩ hoặc chuyên gia y tế để nhận được hướng dẫn phù hợp với tình trạng sức khỏe của bạn.