
Bệnh Tangier là một rối loạn lipid máu di truyền vô cùng hiếm gặp, gây ra bởi sự thiếu hụt trầm trọng hoặc gần như không có lipoprotein mật độ cao (HDL – hay còn gọi là cholesterol “tốt”) trong máu. Tình trạng này dẫn đến hàng loạt bất thường trong vận chuyển cholesterol nội bào và có thể gây tổn thương thần kinh, gan lách to, và các biến chứng tim mạch nghiêm trọng.

Tuy hiếm gặp nhưng việc nhận diện sớm bệnh Tangier có thể giúp kiểm soát biến chứng và hỗ trợ điều trị. Bài viết dưới đây từ ThuVienBenh.com sẽ cung cấp cho bạn cái nhìn toàn diện, chuyên sâu và cập nhật nhất về căn bệnh này – từ nguyên nhân, triệu chứng đến hướng điều trị.
1. Tổng Quan Về Bệnh Tangier
1.1 Định nghĩa và lịch sử phát hiện
Bệnh Tangier lần đầu tiên được mô tả vào năm 1959 tại đảo Tangier, bang Virginia (Mỹ), nơi hai anh em trong một gia đình được phát hiện có mức HDL gần như bằng 0. Bệnh được đặt tên theo địa danh này, phản ánh tính chất địa phương và độ hiếm của nó.
Đây là một dạng rối loạn lipid máu di truyền lặn, do đột biến ở gen ABCA1 – chịu trách nhiệm vận chuyển cholesterol từ tế bào ra máu để hình thành HDL.
1.2 Câu chuyện thực tế: Cậu bé 5 tuổi với mức HDL gần bằng 0
Trong một nghiên cứu đăng trên Tạp chí Lipid học Quốc tế, một cậu bé 5 tuổi không có tiền sử bệnh tim mạch nhưng lại có các triệu chứng thần kinh rõ rệt. Khi xét nghiệm, các bác sĩ phát hiện mức HDL-c trong máu gần như không thể đo được. Sau nhiều đánh giá chuyên sâu, bé được chẩn đoán mắc bệnh Tangier nhờ vào xét nghiệm gen ABCA1. Đây là ví dụ điển hình cho việc cần nghi ngờ và chẩn đoán bệnh kịp thời dù không có dấu hiệu tim mạch rõ rệt.
1.3 Tên gọi và ý nghĩa địa danh Tangier
“Tangier” là tên của một hòn đảo nhỏ ở Mỹ, nơi lần đầu phát hiện ca bệnh. Không chỉ là địa danh, Tangier còn trở thành biểu tượng trong y học cho một hội chứng hiếm mà nghiêm trọng, mang tính cảnh báo giới chuyên môn về một căn bệnh tưởng chừng không tồn tại.
2. Nguyên Nhân Gây Bệnh Tangier
2.1 Vai trò của gen ABCA1
Gen ABCA1 nằm trên nhiễm sắc thể số 9, mã hóa cho một loại protein vận chuyển cholesterol từ tế bào ra ngoài để tạo thành HDL. Khi gen này bị đột biến, cholesterol bị giữ lại trong tế bào, gây tích tụ tại nhiều cơ quan và làm giảm nặng nề lượng HDL trong huyết thanh.
2.2 Cơ chế bệnh sinh do đột biến gen
- Đột biến ABCA1 → rối loạn vận chuyển cholesterol nội bào
- Cholesterol tích tụ tại tế bào mô → gây phì đại gan, lách, hạch
- Thiếu HDL-c → giảm khả năng loại bỏ cholesterol dư thừa khỏi mạch máu
Do đó, bệnh nhân Tangier dễ bị tổn thương mô, thần kinh ngoại biên, và có nguy cơ tim mạch nếu không được phát hiện sớm.
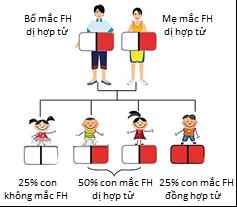
2.3 Tính di truyền trội lặn trên nhiễm sắc thể thường
Bệnh Tangier có kiểu di truyền lặn trên nhiễm sắc thể thường. Điều này có nghĩa là:
- Cả cha và mẹ đều mang gen đột biến mới có khả năng truyền bệnh cho con.
- Người mang một bản sao gen đột biến (carrier) thường không có triệu chứng rõ ràng nhưng có thể truyền cho thế hệ sau.
- Xác suất sinh con mắc bệnh là 25% nếu cả hai cha mẹ cùng là người mang gen đột biến.
3. Biểu Hiện Lâm Sàng Của Bệnh Tangier
3.1 Mức HDL-c rất thấp hoặc không đo được
Đặc điểm sinh hóa nổi bật nhất ở bệnh Tangier là mức HDL-c gần như bằng 0. Đồng thời, tổng cholesterol trong máu cũng thường thấp hơn bình thường. Tuy nhiên, mức LDL có thể dao động và không phản ánh rõ tình trạng bệnh.
3.2 Các triệu chứng đặc trưng
Biểu hiện bệnh phụ thuộc vào mức độ tích tụ cholesterol trong mô và thần kinh:
- Lưỡi vàng cam: do tích tụ cholesterol trong mô dưới niêm mạc
- Hạch bạch huyết to: đặc biệt ở cổ, nách và bẹn
- Gan, lách to: do tích tụ lipid trong mô gan-lách
- Bệnh thần kinh ngoại biên: tê bì, yếu chi, phản xạ giảm
- Suy giảm miễn dịch: dễ nhiễm trùng hô hấp và đường tiêu hóa
3.3 Biến chứng tiềm ẩn
Mặc dù bệnh Tangier thường không gây xơ vữa động mạch như các rối loạn lipid khác, nhưng ở một số bệnh nhân, nguy cơ tim mạch sớm vẫn được ghi nhận. Ngoài ra, các biến chứng thần kinh mạn tính có thể ảnh hưởng nghiêm trọng đến chất lượng cuộc sống nếu không được phát hiện và theo dõi kịp thời.
4. Cơ Chế Bệnh Học: Vai Trò Của ABCA1 Và HDL
4.1 Quá trình hình thành HDL và vai trò ABCA1
HDL được hình thành nhờ quá trình vận chuyển cholesterol từ tế bào mô ngoại vi ra máu, dưới sự hỗ trợ của protein ABCA1. HDL sau đó vận chuyển cholesterol về gan để thải trừ, giúp làm sạch mạch máu và bảo vệ tim mạch.
Hình ảnh: Cơ chế ABCA1 và vai trò trong hình thành HDL
4.2 Rối loạn vận chuyển cholesterol nội bào
Khi ABCA1 không hoạt động, cholesterol bị “mắc kẹt” trong tế bào. Sự tích tụ kéo dài sẽ gây phì đại cơ quan, tổn thương mô và thần kinh. HDL không được tạo thành → cơ thể mất đi công cụ loại bỏ cholesterol dư thừa.
4.3 Tác động toàn thân của sự thiếu hụt HDL
HDL không chỉ là chất vận chuyển cholesterol, mà còn có vai trò chống viêm, chống oxy hóa và bảo vệ nội mô mạch máu. Thiếu HDL kéo dài có thể dẫn đến:
- Suy giảm chức năng mạch máu nhỏ
- Tăng nguy cơ nhiễm trùng
- Rối loạn thần kinh ngoại biên
- Biến chứng tim mạch trong một số trường hợp
Hình ảnh: Sự thiếu hụt HDL có thể gây ảnh hưởng toàn thân
5. Phương Pháp Chẩn Đoán Bệnh Tangier
5.1 Xét nghiệm sinh hóa: HDL, cholesterol toàn phần
Chẩn đoán bệnh Tangier bắt đầu bằng xét nghiệm máu định kỳ. Dấu hiệu nổi bật nhất là HDL-c gần như bằng 0 mg/dL (trong khi mức bình thường là ≥ 40 mg/dL ở nam và ≥ 50 mg/dL ở nữ). Tổng cholesterol cũng thường thấp. Triglycerid có thể tăng nhẹ nhưng không đặc hiệu.
Xét nghiệm lipid máu không đủ để chẩn đoán xác định mà cần kết hợp thêm các dấu hiệu lâm sàng và các phương pháp khác.
5.2 Chẩn đoán hình ảnh: Hạch, gan, lách
Siêu âm bụng và MRI có thể cho thấy gan và lách to, đồng thời phát hiện hạch bạch huyết lớn ở các vùng cổ, nách, hoặc trung thất. Sinh thiết hạch hoặc mô lưỡi có thể phát hiện tích tụ cholesterol dạng bọt – một dấu hiệu điển hình ở bệnh Tangier.
5.3 Xét nghiệm di truyền: phát hiện đột biến gen ABCA1
Xét nghiệm gen là tiêu chuẩn vàng để chẩn đoán xác định. Phân tích DNA giúp phát hiện đột biến trên gen ABCA1 – yếu tố gây bệnh cốt lõi. Việc xét nghiệm di truyền còn hỗ trợ sàng lọc tiền hôn nhân và phát hiện người mang gen bệnh (carrier).
Một số trung tâm di truyền hiện nay có thể thực hiện giải trình tự gen thế hệ mới (NGS) để đánh giá toàn diện hơn.
6. Điều Trị Bệnh Tangier: Có Thể Khỏi Hoàn Toàn Không?
6.1 Nguyên tắc điều trị
Hiện nay, chưa có phương pháp điều trị đặc hiệu cho bệnh Tangier. Mục tiêu điều trị là kiểm soát triệu chứng, hạn chế biến chứng, và cải thiện chất lượng sống.
Các chiến lược điều trị chính bao gồm:
- Giảm cholesterol tích tụ tại mô
- Hỗ trợ hệ thần kinh và miễn dịch
- Theo dõi chức năng gan, tim mạch định kỳ
6.2 Biện pháp hỗ trợ: dinh dưỡng, điều chỉnh rối loạn lipid
Chế độ ăn giàu chất xơ, ít chất béo bão hòa và tránh trans-fat có thể hỗ trợ kiểm soát lipid. Tăng cường vitamin E, omega-3 và chất chống oxy hóa giúp bảo vệ tế bào khỏi tổn thương do tích tụ cholesterol.
Statin hoặc fibrat thường không hiệu quả do HDL quá thấp, nhưng một số nghiên cứu nhỏ gợi ý rằng niacin liều thấp có thể tăng nhẹ mức HDL ở người bệnh Tangier, tuy nhiên còn cần thêm dữ liệu lâm sàng.
6.3 Nghiên cứu gen liệu pháp trong tương lai
Gen liệu pháp đang là hướng nghiên cứu tiềm năng để khôi phục chức năng của gen ABCA1. Các thử nghiệm ban đầu trên động vật cho kết quả khả quan, tuy nhiên vẫn còn xa mới ứng dụng được trên người. Ghép tủy cũng từng được đề cập trong một số nghiên cứu nhỏ nhưng chưa chứng minh được hiệu quả dài hạn.
7. Phân Biệt Bệnh Tangier Với Các Rối Loạn Lipid Khác
| Đặc điểm | Bệnh Tangier | Tăng cholesterol di truyền | Bệnh Niemann-Pick type C |
|---|---|---|---|
| HDL | Gần bằng 0 | Bình thường hoặc tăng | Thường bình thường |
| LDL | Giảm nhẹ hoặc bình thường | Tăng rất cao | Bình thường hoặc tăng |
| Triệu chứng thần kinh | Rõ rệt, mạn tính | Không có | Có, tiến triển nặng dần |
| Di truyền | Lặn NST thường (ABCA1) | Trội (LDLR, APOB…) | Lặn NST thường (NPC1/NPC2) |
8. Tầm Quan Trọng Của Phát Hiện Sớm Và Tư Vấn Di Truyền
8.1 Sàng lọc tiền hôn nhân và tiền sinh
Với các bệnh di truyền hiếm như Tangier, sàng lọc di truyền đóng vai trò cực kỳ quan trọng. Việc xét nghiệm gen trước khi kết hôn giúp phát hiện những người mang gen đột biến, từ đó tư vấn nguy cơ sinh con mắc bệnh.
8.2 Tư vấn cho gia đình mang gen bệnh
Người trong gia đình bệnh nhân nên được kiểm tra để phát hiện người mang gen lặn (carrier). Dù họ không biểu hiện triệu chứng, nhưng có thể truyền gen sang thế hệ sau. Tư vấn di truyền đóng vai trò then chốt trong việc ngăn ngừa tái phát bệnh trong gia đình và cộng đồng.
9. Kết Luận
9.1 Bệnh hiếm nhưng cần cảnh giác
Bệnh Tangier là rối loạn lipid di truyền hiếm gặp nhưng có thể gây biến chứng nghiêm trọng nếu không được phát hiện kịp thời. Việc nhận biết các triệu chứng như HDL thấp bất thường, lưỡi vàng cam, hạch to và tổn thương thần kinh là dấu hiệu quan trọng để tiến hành xét nghiệm sâu hơn.
9.2 Hướng nghiên cứu tương lai và hy vọng điều trị
Dù chưa có cách điều trị khỏi hoàn toàn, nhưng nhờ vào sự tiến bộ của y học và công nghệ gen, tương lai điều trị cho bệnh Tangier đang mở ra nhiều cơ hội. Sự phối hợp giữa bác sĩ lâm sàng, nhà di truyền học và gia đình bệnh nhân là yếu tố quyết định trong quản lý và theo dõi bệnh hiệu quả.
“Chẩn đoán đúng là bước đầu tiên quan trọng nhất trong cuộc hành trình sống khỏe cùng bệnh di truyền.” – BS. Peter T. Lansberg, chuyên gia lipid học – Đại học Groningen
FAQ – Các Câu Hỏi Thường Gặp Về Bệnh Tangier
1. Bệnh Tangier có nguy hiểm không?
Có, vì nếu không được chẩn đoán và theo dõi, bệnh có thể gây tổn thương thần kinh mạn tính, suy giảm miễn dịch, và trong một số trường hợp là biến chứng tim mạch.
2. Bệnh Tangier có di truyền không?
Có. Đây là bệnh di truyền lặn trên nhiễm sắc thể thường, cần cả cha và mẹ đều mang gen đột biến để con có nguy cơ mắc bệnh.
3. Có xét nghiệm nào phát hiện bệnh sớm không?
Có. Xét nghiệm lipid máu cho thấy HDL rất thấp là dấu hiệu đầu tiên. Xác nhận bằng xét nghiệm di truyền gen ABCA1.
4. Người mang gen bệnh (carrier) có triệu chứng không?
Thường không có triệu chứng rõ ràng, nhưng có thể có mức HDL thấp nhẹ. Vẫn nên theo dõi sức khỏe định kỳ và tư vấn di truyền trước khi có con.
5. Có điều trị khỏi hoàn toàn được không?
Hiện chưa có thuốc điều trị đặc hiệu. Điều trị chủ yếu là kiểm soát triệu chứng, cải thiện chất lượng sống và phòng biến chứng.
📝Nguồn tài liệu: Chọn lọc từ nhiều nguồn y tế uy tín
🔎Lưu ý: Bài viết chỉ nhằm mục đích cung cấp thông tin tổng quan. Vui lòng tham khảo ý kiến của Bác sĩ, Dược sĩ hoặc chuyên gia y tế để nhận được hướng dẫn phù hợp với tình trạng sức khỏe của bạn.